在后基因組時代,蛋白質組學在生物醫學研究中發揮著重要作用。2022年8月,Nature子刊《Laboratory Investigation》發表了一篇高通量蛋白組的mini-review,概述了高通量蛋白質組學技術、統計和算法的進展。

蛋白質組學作為蛋白質組實驗和數據分析的結合,從整體上分析了蛋白質的組成、結構、表達、修飾狀態以及蛋白質之間的相互作用和聯系。它為基因組學和轉錄組學提供補充信息。它對于生成復雜的、相互關聯的通路、網絡和分子系統的圖譜也是至關重要的,這些通路、網絡和分子系統直接控制著主要的生命活動,如細胞的增殖、分化、衰老和凋亡。隨著過去十年實驗技術的大幅提高,蛋白質組學方法已經從傳統的免疫組織化學(IHC)染色、western blot和酶聯免疫吸附試驗(ELISA),發展到高通量的方法,如組織微陣列(TMA)、蛋白質通路陣列和質譜分析。這些高通量蛋白質組學技術不僅減少了分析時間,還提高了蛋白質組覆蓋的準確性和深度。隨著生物信息學和現代多分析 "組學 "技術的發展,蛋白質組學在揭示疾病的分子機制,發現新的生物標志物方面有著巨大的前景,并可作為特定的診斷檢測、預后預測和治療目標,進一步加強個性化醫療。

蛋白質組學“從實驗到臨床”
質譜分析(Mass spectrometry)
質譜(MS)已經發展成為識別蛋白質及其異構體的最基本和最流行的工具之一,并通過直接的片段或負責其形成的特定蛋白質分解活動對翻譯后修飾進行量化。MS最重要的作用是發現和檢測一個完整的蛋白質或復合肽或代用肽的子集。MS可以與多種分離和預分餾技術相結合,以識別目標蛋白/肽,提高識別的準確性和產量。例如2D-PAGE基于電荷和分子量,而液相色譜(LC)基于極性、電荷和蛋白質分子量。盡管2D-PAGE傳統上被用作蛋白質組學研究的標準程序,但基于凝膠的技術往往是勞動密集型和耗時的,因此不適合于高通量蛋白質組學。相比之下,液相色譜或高效液相色譜(HPLC)可以從復雜的混合物中連續分離數千種蛋白質,并可與質譜結合成為LC-MS,以提高通量。其中,反相液相色譜法(RPLC)是最常用的基于LC的分離平臺。
根據不同的處理策略,基于質譜的方法可分為自上而下、自下而上和鳥槍法。在自上而下的蛋白質組學方法中,全長的蛋白質可以隨后在質譜內被分割,并記錄片段的質量,直接被送去進行質譜分析。相比之下,在自下而上的蛋白質組學技術中,蛋白質被酶解或化學消化成肽輸入到質譜設備中。此外,鳥槍蛋白質組學是自下而上蛋白質組學的一個特殊案例,復雜混合物(如血清、尿液和細胞裂解液)中的整個蛋白質被切割成肽,然后進行多維HPLC-MS,其目的是像基因組 "鳥槍 "測序一樣產生蛋白質混合物的全局概況。另一方面,在自下而上的策略中,不一定需要在MS之前分離多肽。然后,通過依賴數據的發現引擎,對MS數據進行匹配,以確定蛋白質序列數據庫中的目標蛋白質及其相關修飾,這可分為肽段評分、蛋白質評分,最后是蛋白質推斷。
蛋白通路芯片(Protein pathway array)
蛋白質通路陣列(PPA)是一個基于凝膠的高通量平臺,采用抗體混合物來檢測蛋白質樣品中的抗原,這些樣品可以從活檢或組織中提取。在這種方法中,可以對腫瘤組織進行顯微切割,以最大限度地提高來自腫瘤組織而不是周圍良性組織的蛋白質的比例。然后,將抗體-抗原反應的免疫熒光信號轉換為數值數據,作為蛋白質表達的數值。在數據歸一化和適當的統計學建模后,可以探索和訓練生物標志物和蛋白質組網絡。PPA已被應用于許多疾病,如原發性血小板增多癥和甲狀腺乳頭狀癌。其高通量的蛋白質譜系以穩健的定量方式提供了比傳統方法更多的優勢。
高通量組織芯片(Next generation tissue microarrays)
隨著過去十年技術的進步和高通量需求的增加,TMA逐漸開始在研究和臨床領域廣泛使用。TMA包含了數百個不同病例的福爾馬林固定石蠟包埋(FFPE)或冷凍塊的許多小的代表性組織,以陣列方式組裝在一張組織學玻片上,因此可以同時對多個樣本進行大規模的基于抗體的分子分析。因此,它是確認和驗證由PPA或MS蛋白質組學方法產生的新生物標志物的一個實用和有價值的工具。因此,它經常被用于獨立的隊列,并確定目標蛋白在細胞膜、細胞質或細胞核中的位置。由于在過去的一年里,具有多種智能顯微鏡的數字病理學得到了快速發展,最近創建了一種新的TMA方法,即高通量組織微陣列(ngTMAs)。它允許將注釋直接放在數字玻片上,以獲得更高的準確性。ngTMAs的兩個主要優點是其時間效率和高通量而不影響質量。由于其改進的靈敏度和快速、大規模的檢測能力,ngTMAs已成為提高臨床和轉化研究中使用的TMA質量的有力工具。
基于磁珠或適體的多重檢測(Multiplex bead- or aptamer-based assays)
近年來,基于Luminex珠陣列系統越來越多地被用于蛋白質分析應用中。它使蛋白質組學生物標志物panel的檢測可靠、快速,并能應對各種臨床實踐中的動態變化。另一個廣泛使用的平臺是Meso-scale Discovery (MSD),對于人類細胞因子譜,MSD測定比Luminex測定更敏感,但特異性較低。Simoa?是廣泛使用的bead-based的多重檢測之一。它涵蓋6個疾病領域,可定制,截至2022年5月,包括109個腫瘤學、26個神經學、19個免疫學、13個心臟病學和45個傳染病檢測。基于適體的SOMAscan?分析可在單個檢測中評估1000至9000種抗原的表達,并具有廣泛的動態范圍(8個數量級)、極高的靈敏度(檢測下限,40fm)和高精度(中位變異系數=5%)。
鄰位延伸技術(Proximity extension assay (Olink)
PEA作為鄰近依賴性連接分析之一,基于彼此具有輕微親和力的寡核苷酸連接抗體對。當這些寡核苷酸連接的抗體接近時,與抗體連接的兩個獨特的寡核苷酸將被DNA聚合酶延伸,并在隨后以指數方式擴增。Olink檢測法已經成功應用于多個臨床領域,包括2019年冠狀病毒疾病(COVID-19)、創傷性腦損傷和腎臟疾病。它可以在極少量的樣品(如幾微升)中同時對3000多種蛋白質進行定量。
基于納米孔的單分子蛋白質組學(Nanopore based single-molecule proteomics
納米孔已越來越多地用于DNA或RNA測序,并嘗試用于蛋白質組學。它在蛋白質組學的早期應用是在分枝桿菌肽的測序中。基于納米孔的單分子蛋白質組學的主要挑戰是檢測翻譯后修飾(PTM)的效率低和缺乏足夠的靈敏度。
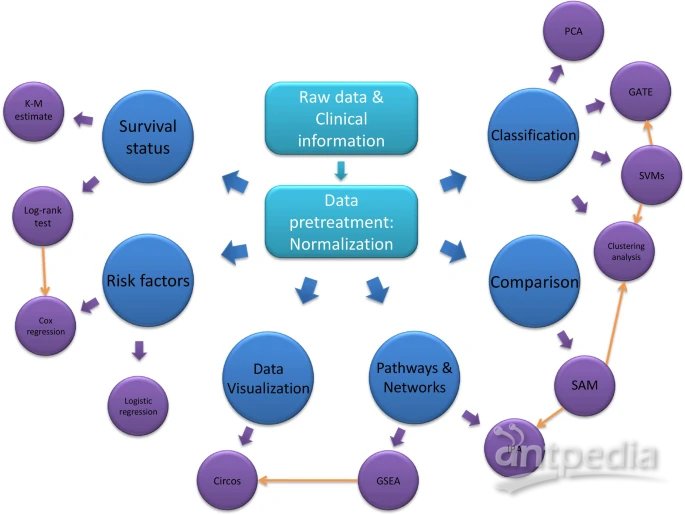
歸一化
大數據預處理階段最常見和必要的形式是歸一化,用于將所有數據集中并重新縮放為一個整體數值矩陣,以提高其數值穩定性、整體性能和模型擬合。例如,最常用的歸一化公式是Z-score,也稱為標準分數。
芯片顯著性分析(SAM)
SAM是一個Microsoft Excel插件包,是一種廣泛使用的基于高通量的互換方法,使用改進的t-statistics (q-value)來識別在蛋白質組數據中多組樣本之間的差異表達蛋白,該方法衡量蛋白質豐度和疾病結果之間關系的強度。與小樣本量的常規t-test不同,SAM算法非常適用于大數據,通過對蛋白質豐度的列進行置換,并通過最近鄰算法自動插補缺失數據,使假陽性和假陰性的數量最小化。此外,SAM的一個有價值的特點是,它利用數據的排列組合給出了錯誤發現率的估計值,即可能被偶然確定為有意義的蛋白質的比例。

SAM分析生成的圖
聚類和判別分析
層次聚類算法(HCA)已被用于通過形成基于數學模型的樹狀圖對大數據進行聚類。為了測量數據點之間的距離,建立了幾個基于數學公式的優化模型,包括曼哈頓距離(L1)、歐幾里得距離(L2)、皮爾遜相關系數等。距離度量的選擇會影響HCA的性能,因此應謹慎決定。在HCA之前,應明確定義基本變量(生物標志物)、樣本選擇標準和研究目標,以便進行穩健和可重復的分析。此外,HCA可分為單向和雙向HCA。還有一種特殊的聚類分析,稱為時間序列表達的網格分析(GATE),用于根據時間序列分析和可視化高維生物分子。

層次聚類分析(HCA)和時間序列表達的網格分析(GATE)示例
與對已知樣本進行分類的聚類分析不同,(預測性)判別分析是根據算法在訓練集中學習和建立的內容對未知樣本進行分類,例如不依賴于數據類型的支持向量機(SVM)可用于線性分離數值或分類數據,并確定潛在的生物標志物作為分類器。(基于機器學習與否)判別分析的主要目的是設計一個計算有效的統計學模型,對多組受試者進行分類,并確定預測率較高的潛在分類器。
生存分析
Kaplan-Meier(K-M)曲線是一種時間事件統計方法,用于研究終點事件與時間周期之間的關系。它可用于評估生存時間、疾病復發、臨床試驗、動物研究等。K-M估計是計算生存時間的最簡單方法。兩條生存曲線可以通過log-rank (Mann–Whitney U) 檢驗進行統計比較,該檢驗已被廣泛使用,包括計算過程中具有不同權重函數的Breslow和Tarone。但它們通常作為單變量分析,不允許測試其他疾病相關變量的影響。相比之下,經常用作多變量分析的Cox比例風險回歸模型可以在識別疾病自變量的同時測試其他變量的影響。此外,許多流行的回歸模型被用于分析蛋白質組學或基于芯片的大數據,它們的功能在不同程度上類似于Cox回歸模型,例如多變量邏輯回歸。
主成分分析
主成分分析(PCA)的主要目的是通過創建一組稱為主成分的新變量來降低大數據的維數,以表示原始數據集中的大部分信息。因此,只有前幾個主成分是最有代表性的,這種每個主成分的變異性逐漸減少的趨勢可以用scree圖來表示。這種通過主成分降低數據集內維度表示的統計方法對于大數據集或大數據的分類和壓縮非常有用。

主成分分析
Ingenuity通路分析、基因集富集分析和circos分析
許多分析方法結合在線數據庫分析蛋白質組學和微陣列數據,更適合于發現臨床意義。Ingenuity通路分析(IPA)是一種基于web的軟件應用程序,用于使用表達式數據集進行因果分析。IPA可以同時可視化和分析基因組學、蛋白質組學和代謝組學數據的跨數據庫數據,以獲得綜合各種組學格式的信號網絡和典型通路。基因集富集分析(Gene set enrichment analysis, GSEA)是另一種計算方法提供通路富集工具來幫助解釋數據集。這種方法關注多個基因作為基因集表達的累積變化,這些基因集共享相似的生物功能、染色體位置或調控,而不是單個基因來識別通路。GSEA方法最顯著的優點是它可以捕捉到一些通路,其中幾個基因在少量但以一種協調的方式改變。

IPA和GSEA分析示例
此外,Circos是一個軟件包,用于在圓形布局中可視化基于組學的數據和信息。可以創建Circos圖來探索經典通路與臨床病理特征或風險因素之間的關系和貢獻。

Circos圖示例:在所有八種臨床病理類型中,性別占分布的最顯著比例,表明是對信號網絡影響最大的臨床因素。
單細胞蛋白組學
單細胞蛋白質組學是一種新興的單細胞技術。在不久的將來,它將競爭和補充單細胞轉錄組學,以了解單細胞生物學。單細胞蛋白質組學最近成為現實,因為先進的技術表明,單細胞中的肽可以有效地輸送到MS儀器。這些單細胞質譜方法可廣泛分為無細胞和多重方法,后者允許同時對多個細胞進行蛋白質組學分析。SCoPE2和Scp是用于分析多重單細胞蛋白質組數據的R包,而SCeptre是它們在Python中實現的對應軟件包。一些通用的蛋白質組學流程也可用于處理單細胞蛋白質組學數據,包括計算質量控制工具和用于數據處理和可視化的單一流程(MSnbase)。
總之,在過去的十年里,蛋白質組學技術和研究取得了巨大的進步。高通量蛋白質組學方法的能力不斷增強,已經產生了實時和深入的數據集。有效的數據挖掘技術也大大有助于尋找新的有用的生物標志物,這對疾病的早期檢測和治療至關重要。隨著計算能力的突破和人工智能的興起,蛋白質組學的作用進一步擴大。高度先進的統計/計算模型使蛋白質組學能夠整合到多組學中。在這一新趨勢下,蛋白質組學數據分析將發生革命性的變化,以獲得大量臨床和健康相關數據的更大藍圖。這是蛋白質組學發展成為一門重要的新學科并與其他學科融合的激動人心的時刻。盡管蛋白質組學在這一過程中面臨著新的挑戰,但它將朝著更深入的單細胞生物學和個體化精準醫學的方向發展,將基礎研究和臨床實踐提升到另一個水平。
由于篇幅有限,更多技術細節可參考文獻原文:https://www.nature.com/articles/s41374-022-00830-7
參考文獻
Cui M, Cheng C, Zhang L. High-throughput proteomics: a methodological mini-review[J]. Laboratory Investigation, 2022: 1-12.
19世紀中葉,瑞士人類學家巴霍芬在《母權論》(1861)一書中首次提出人類社會的童年曾普遍存在一個母系社會的發展階段,但這缺乏考古學上支持史前母系社會存在的有力證據。另一方面,現代民族學研究所揭示的母......
作為當前生物醫學研究的前沿熱點領域,單細胞蛋白質組學通過在單細胞層面上探索蛋白質表達模式,精確揭示細胞異質性,為解碼生命過程和疾病發生機制提供了獨特視角,已成為賦能精準醫療和生物醫藥創新的關鍵技術,是......
酶聯免疫檢測儀(簡稱酶標儀),又稱微孔板檢測器,是現代生物醫學研究與臨床檢驗領域中不可或缺的分析儀器,主要用于酶聯免疫吸附試驗(ELISA)。它通過檢測酶促反應產生的光信號來定量分析樣本中的特定物質,......
基因測序儀作為生命科學研究和醫學應用的關鍵工具,通過對核酸分子的序列測定,為遺傳信息的解讀和挖掘提供了基礎。自第一代測序技術誕生以來,測序儀經歷了從一代到三代的快速發展。一代測序儀主要基于Sanger......
《自然》近日揭曉了21世紀被引用次數最多的25篇論文。令人意外的是,mRNA疫苗、CRISPR基因編輯、希格斯玻色子的發現等重大突破性成果均未進入榜單。真正入榜的,反而是涉及人工智能(AI)、提升研究......
為推動生物質譜與功能組學領域的深度交流與協同創新,香港中醫藥表型組學研究中心攜手SCIEX中國和功能代謝組科學實驗室,聯合打造高端學術平臺——紫金花論壇。論壇聚焦交叉學科研究前沿,鏈接全球學術與產業智......
你是否曾經歷過這樣的場景?某次不小心誤食了變質的海鮮,結果上吐下瀉,之后哪怕只是看到類似的食物,甚至聞到一絲相關氣味,都會感到強烈的惡心和不適。這種“一朝被蛇咬,十年怕井繩”的現象,其實是大腦中一種深......
多重耐藥致病真菌的全球傳播對人類健康構成了嚴重威脅,因此有必要發現具有獨特作用模式的抗真菌藥物。然而,由于已知化合物的高頻率重新發現和缺乏新的抗真菌藥物靶點,傳統的基于活性的篩選先前未描述的抗生素受到......
“超乎想象!”《自然》(Nature)審稿人在論文評審意見中說。中國科學院天津工業生物技術研究所研究員高書山告訴《中國科學報》:“他們(指審稿人)大概都覺得太顛覆了,給我們的評審意見都是這樣的調調:你......
由于無法對蛋白實現擴增,因此在單細胞多組學的研究中,單細胞蛋白質組學是最具挑戰的研究,近年來已取得突飛猛進的進展。除了質譜儀檢測靈敏度的提升,在前端取樣、預處理和分離方面的巨大進步是重要因素,這和微流......